Dear users, please have a look at the introductory video here. Alternatively you can go to the feature videos section from the drop-down menu at the top left corner. You can watch recent updates and features added to i-PV from here.
You might also want to try a sample output by clicking on the top left ![]() icon.
icon.
For people who want to download the program, the links will be opened soon.. Installation/usage process and the walkthroughs will also be updated.
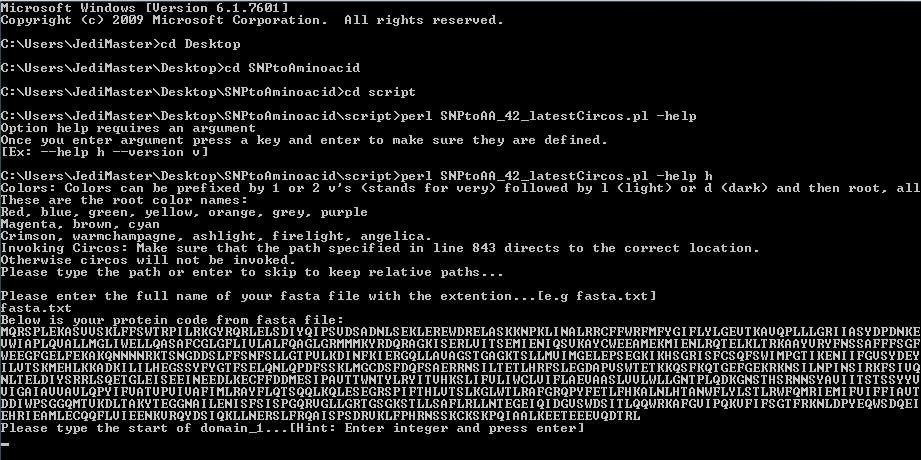
Here I will demonstrate a short tutorial for how to use I-PV.
Before we begin, you will need 4 files:
-fasta file (ex. fasta.txt)
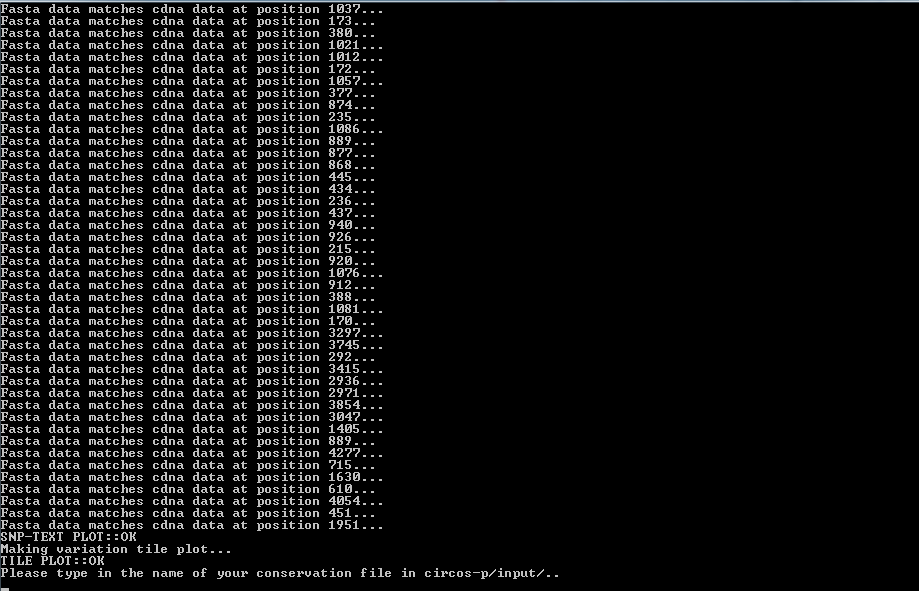
-conservation file (ex. conservation.txt). This is just a text file with 1 number per line. If you wish to skip this step and use the outer track as a domain label just provide
a casual tab delimited text file with a single number per line. You can always change the histogram by uploading files to your i-pv output using the icons in the lower right corner.
-mRNA file (ex. mRNA.txt)
-Mutation file (ex. biomart.txt). You can easily download this for your transcrip of interest from Biomart.
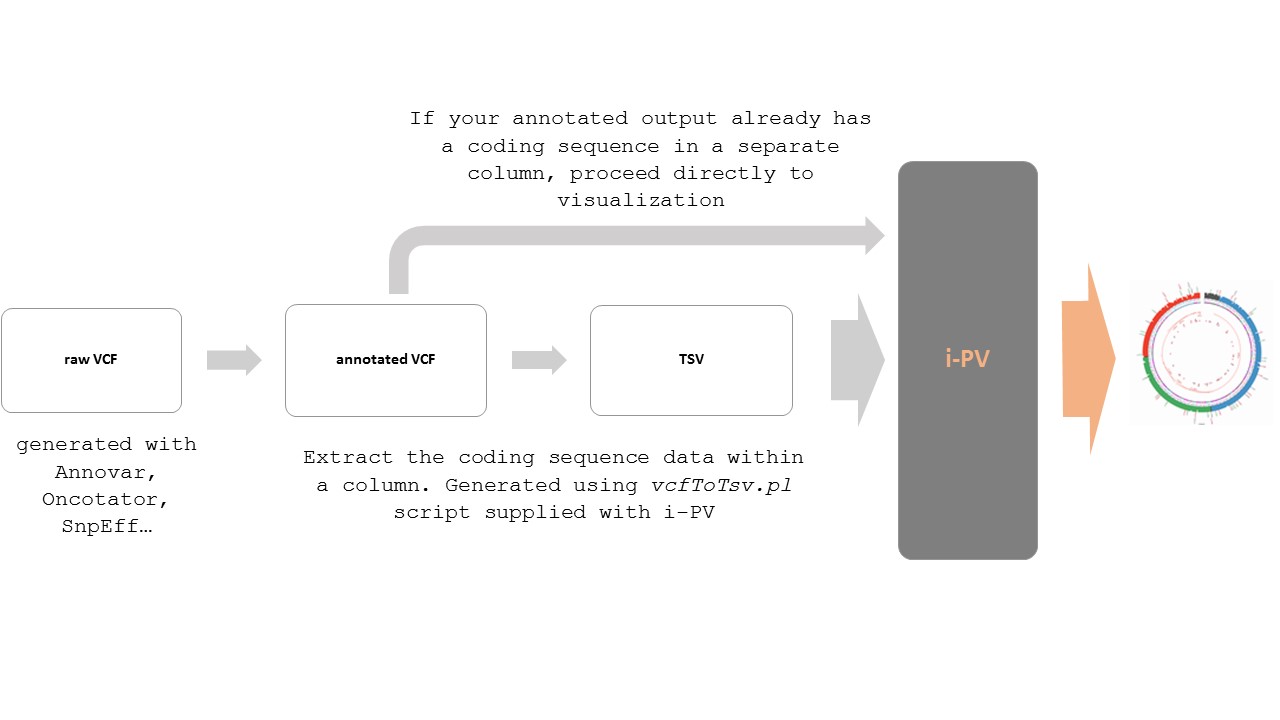
Alternatively, if you have a custom vcf file you first have to annotate it via either ANNOVAR, Snpeff,
Oncotator or other commercially available vcf tools. I-PV uses the coding sequence and checks if variants reported in your annotated vcf match correctly to your fasta files.
Therefore you need to make sure that your annotated VCF files contain the coding sequence start (CDS) of the variation. This is generally expressed in the format c.XXXA>T for SNPs and c.XXX_YYYdelinsAATT.. for indels. If your annotated files have a single column containing only the CDS then proceed to visualization and plotting.
If your CDS values are embedded in a column with other metadata then you need to extract this CDS information and make a new i-PV compatible TSV file using the vcfToTsv.pl script.The general scheme to follow with VCF files are shown below:
First start by opening the command line and navigationg to the I-PV/script folder.
After opening the command line you will be asked to provide the fasta file.
The fasta file does not need any preformatting..
After you provide the fasta file you will need to provide the mRNA file.
It will be subsequently matched to your fasta. You will get an error message if non of the possible frames
fit your fasta file..
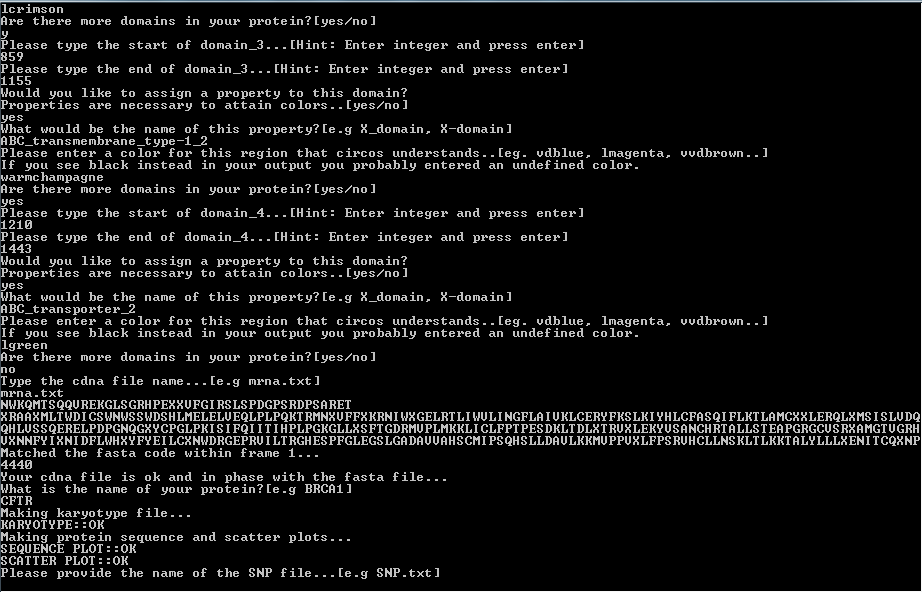
Next you will have to provide the mutation file
Follow the on screen instructions carefully and don't
forget that countinf the columns start from 1.